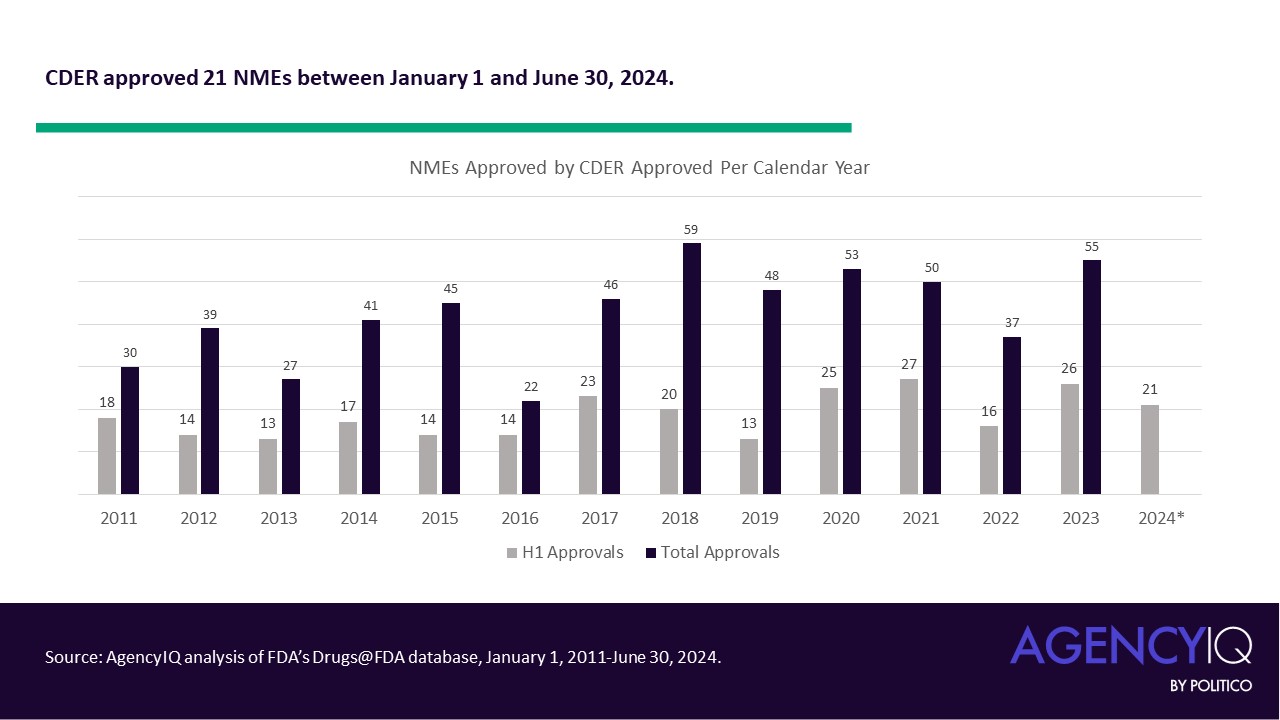
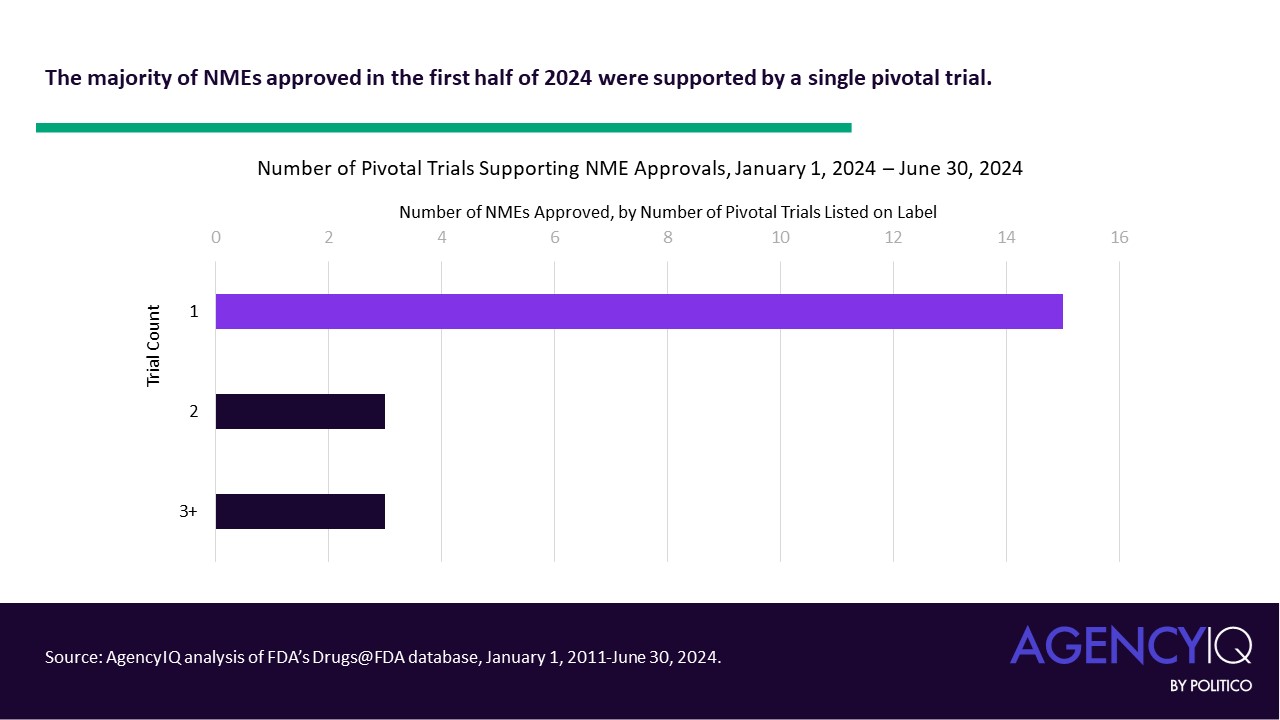
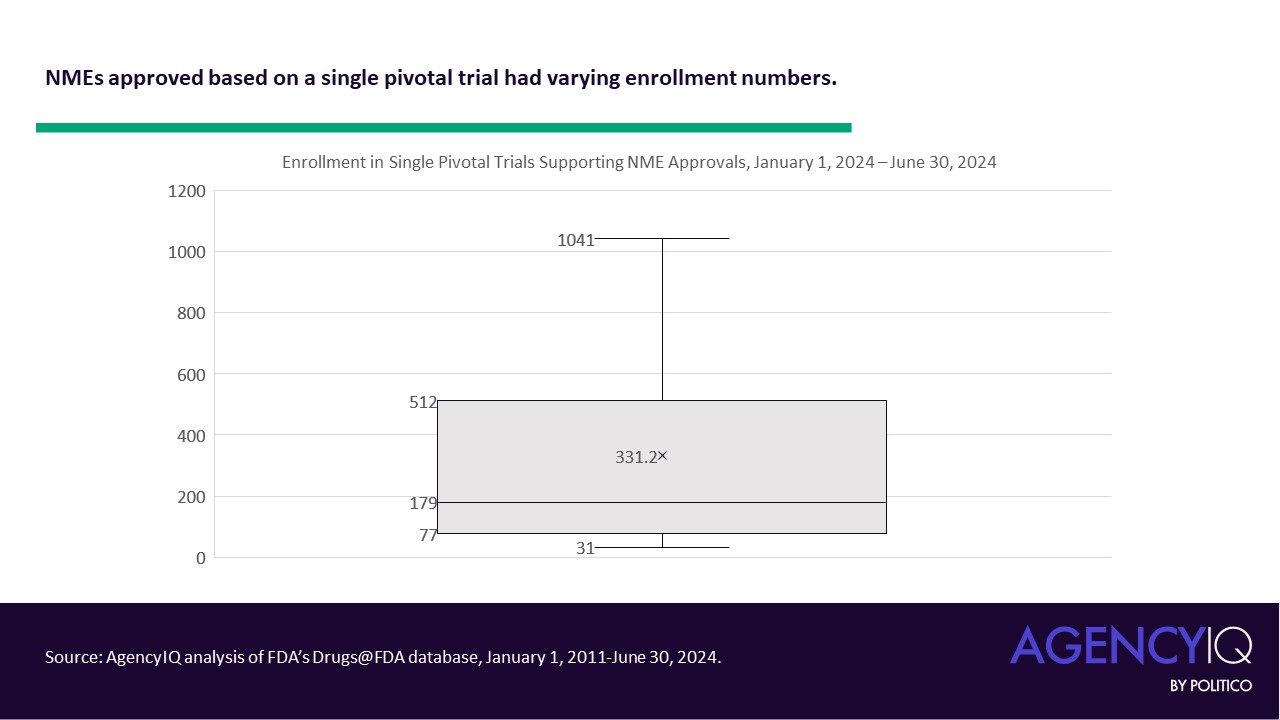
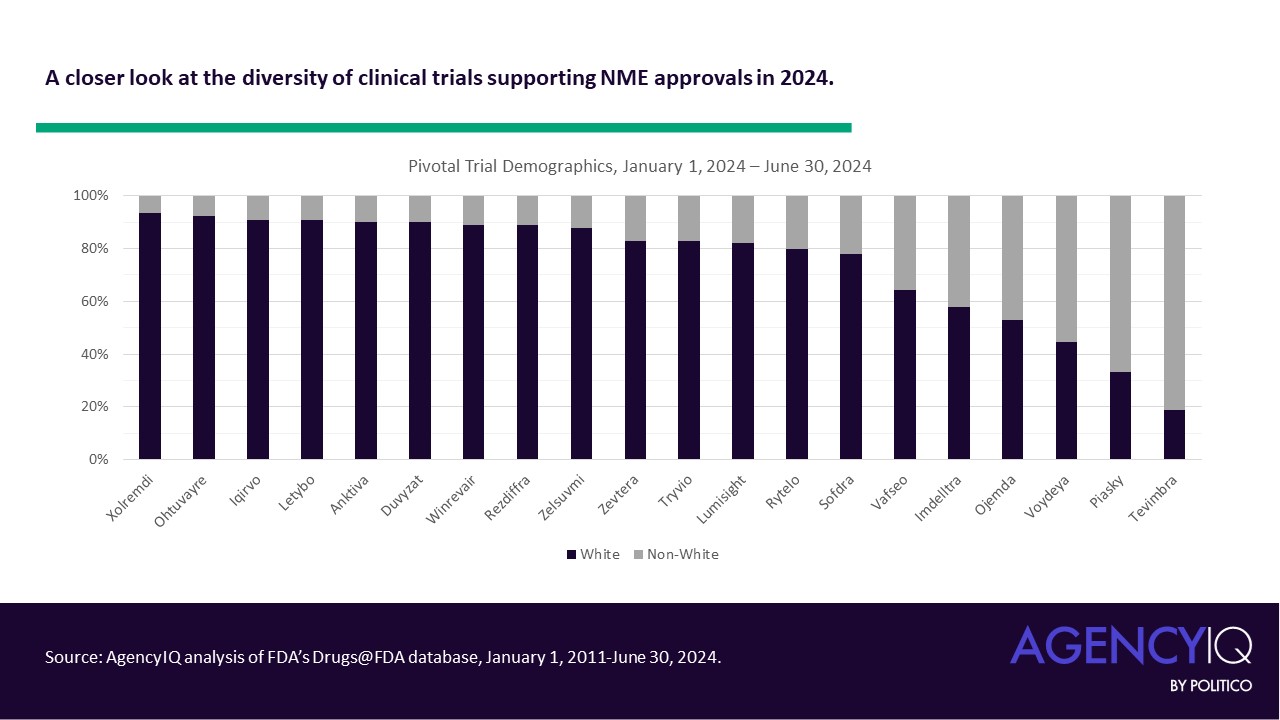
The new EUDAMED roadmap includes updates accounting for the recent amendments to the European medical device and diagnostics regulations, which allow for a phased rollout of the different functional modules of the database. However, it’s still not clear when the clinical investigation and performance study module will be done.
Fill out the form to read the full article.